
This story has been updated with additional information provided by the Food and Drug Administration after original publication.
The company at the center of a worldwide sleep aid device recall that left many sleep apnea patients feeling restless is now under scrutiny by federal investigators on several fronts.
The Food and Drug Administration (FDA) announced it has received more than 100 reports of deaths associated with popular sleep aid devices many relied on worldwide.
This latest revelation comes after the company behind that recalled product - Philips Respironics, a subsidiary of the company Royal Philips - recently informed shareholders it was served subpoenas by the U.S. Department of Justice, as part of an ongoing investigation into the Respironics recall.
The worldwide voluntary recall by Philips of CPAP, BiPAP and ventilator devices was first announced nearly a year ago in June of 2021 after the company said there was a noise-abatement carcinogenic foam inside the device that could breakdown, and be unknowingly inhaled by its user.

The FDA quickly labeled the recall as a Class One: a designation reserved for the most serious kind of recall.
FDA investigators later reported the recall impacted more than 15 million devices worldwide.
So many sleep apnea patients across the globe relied on the devices covered under the recall that Philips said it did not have enough parts to repair or replace devices immediately, leaving some patients without many options.
Patients like Edward Coleman, a 73-year-old Vietnam veteran out of Michigan City, Indiana, who said his doctors at the VA prescribed him a Philips DreamStation for his sleep apnea, regulating his breathing while he sleeps.
Feeling out of the loop? We'll catch you up on the Chicago news you need to know. Sign up for the weekly Chicago Catch-Up newsletter.
“My sleep is interrupted, you know, between two-to-three times a night,” Edward explained. “So, it's hard to really get a good night's rest.”
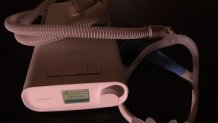
Coleman said he’s still waiting on a replacement device, and since learning of the recall, he’s now sleeping without any sleep-aid device after weighing the risks, but he worries for other veterans out there who may not know about the problem.
“I want to reach out to the other veterans out there so that they may get the information that they need to have their machines repaired or replaced,” Coleman told NBC 5 Responds. “That's my utmost concern at this point.”
FDA: 124 Suspected Deaths From Philips Devices
Coleman’s frustration, like many Americans, isn’t dulled by the latest revelations from regulators.
Since April 2021, the FDA said it has received “more than 21,000 medical device reports (MDRs)” associated with the breakdown of the noise-abatement foam inside Philips Respironics ventilator, BiPAP and CPAP devices.
Those MDRs include 124 reports of deaths associated with Philips' recalled devices, the FDA said.
MDRs are a post-market surveillance tool used by the FDA to monitor medical devices, including reports of device-associated malfunctions, injuries or deaths.
These MDRs to the FDA include both mandatory reports from Philips as well as voluntary reports from health professionals, consumers, and patients, an FDA spokesperson said.
The FDA told NBC 5 Responds it is looking into each report, but felt it was important to share the preliminary findings with the public.
“We are in the process of evaluating each report while communicating this early information to public. There is, based solely on the reports, no immediate causal link between the deaths and the foam breakdown,” an FDA spokesperson said. “The 124 deaths have not been verified beyond the medical device reports submitted to the FDA.”
In response to the FDA’s findings, Philips told NBC 5 Responds that while the company “investigates all allegations of device malfunction, death, or serious injury… the cause of an event cannot typically be determined from [the FDA’s MDR] reporting system alone.”
“Philips Respironics received a steep increase in complaints allegedly associated with possible foam degradation,” after the company announced the recall in June 2021, a spokesperson told NBC 5 Responds.
The company said, “Philips regrets any inconveniences caused by this issue and we are committed to supporting the community of patients who rely on our sleep and respiratory care solutions.”

The company also pointed to a study published by the American Journal of Respiratory and Critical Care Medicine (AJRCCM) last December that found patients using its devices did not have a higher risk of cancer.
The company said, “Independent of Philips Respironics, in December 2021, an analysis was published in the [AJRCCM] that did not find a higher risk of incident cancer among obstructive sleep apnea (OSA) patients who used a Philips Respironics PAP device as compared to OSA patients who used a PAP device from other manufacturers, or OSA patients without treatment,”
NBC 5 Responds contacted the AJRCCM to verify details about the study but did not hear back by the deadline.
DOJ Subpoena and Lawsuits
Philips is beginning to feel increased pressure brought on by the recall.
In a call last month with shareholders, Royal Philips’ Chief Executive Officer Frans van Houten said the company and its subsidiaries were served subpoenas on April 8 by the U.S. Department of Justice in connection to an ongoing investigation into the recall.
The subpoena from the DOJ was “to provide information related to events leading to the Respironics recall,” van Houten said. “The relevant subsidiaries are cooperating with the agency.”
The CEO added that no specific allegations were raised in the subpoenas, rather it was only a general request for information about the recall.
Pressure is also mounting in courtrooms across the country.
In that same call, the company acknowledged it’s currently facing 185 personal injury lawsuits and more than 100 class-action lawsuits tied to the device recall. Those class-action lawsuits are expected to be combined into two lawsuits this summer, the company said.
NBC 5 Responds reported in December that an FDA inspection found nearly a decade ago, the company received more than a hundred complaints related to the sleep aid device product defects, but did not immediately act.
In a previous statement, the company downplayed those FDA findings, saying of the “limited complaints” it received, each was “evaluated and addressed”, case-by-case.
To read more about the FDA’s inspection of Philips, click here or watch below.
From the moment it announced the recall, Philips said it plans to repair or replace every device impacted, but that it will take time.
To date, out of more than 15 million devices worldwide, the company said it has shipped 1.1 million replacement devices and repair kits to patients across the United States.
In the company's April earnings call, CEO van Houten said it hopes to have 90% of the replacement machines sent out to customers in 2022.
Below is a complete list of the Philips Respironics machines that are impacted, according to the FDA:
CPAP and BiPAP Devices
| Device Type | Model Name and Number (All Serial Numbers) |
| Continuous Ventilator, Minimum Ventilatory Support, Facility Use | E30 (Emergency Use Authorization) |
| Continuous Ventilator, Non-life Supporting | DreamStation ASV, DreamStation ST - AVAPS, SystemOne ASV4, C-Series ASV, C-Series S/T and AVAPS, OmniLab Advanced+ |
| Noncontinuous Ventilator | SystemOne (Q-Series), DreamStation, DreamStation Go, Dorma 400, Dorma 500, REMstar SE Auto |
Ventilators
| Device Type | Model Name and Number (All Serial Numbers) |
| Continuous Ventilator | Trilogy 100, Trilogy 200, Garbin Plus, Aeris, LifeVent |
| Continuous Ventilator, Minimum Ventilatory Support, Facility Use | A-Series BiPAP Hybrid A30 (not marketed in the US), A-Series BiPAP V30 Auto |
| Continuous Ventilator, Non-life Supporting | A-Series BiPAP A40, A-Series BiPAP A30 |

